过度金属催化的碳氢键活化-官能团化反应是一个非常高效、高原子经济性的构建新碳碳键、碳杂原子键的合成手段。在最近的十年期间,大量实用的铑催化碳氢键活化-官能团化反应被广泛报道。与此同时,针对这些反应机理的理论和实验研究也成为国内外化学家关注的焦点之一。

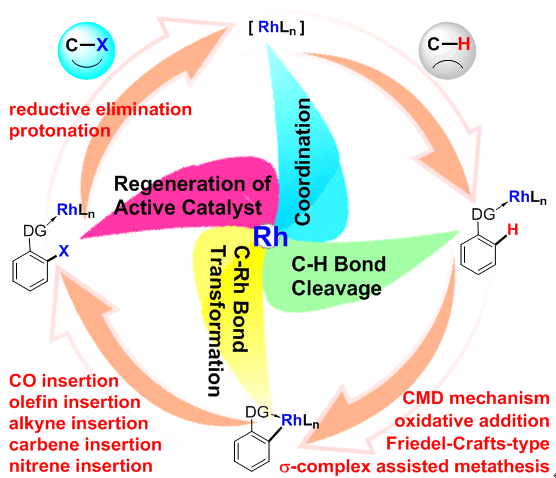
自从2012年开始,蓝宇教授课题组就在铑催化碳氢键活化-官能团化反应机理研究方面展开了系统、深入地探索。研究内容包括铑催化的碳氢键活化-卡宾插入反应(J. Am. Chem. Soc. 2015, 137, 1623.)、碳氢键活化-炔烃插入反应(Chem. Eur. J. 2015, 21, 10131; J. Phys. Chem. A 2017, 121, 4496; ACS Catal. 2017, 7, 7296; J. Am. Chem. Soc. 2017, 139, 15724.)、碳氢键活化-氮宾插入反应(ACS Catal. 2016, 6, 1971; Angew. Chem. Int. Ed. 2016, 55, 8696.)、碳氢键活化-羰基插入反应(Organo llics 2016, 35, 1480.)、碳氢键活化-烯烃插入反应(ACS Catal. 2016, 6, 7744.)、选择性sp3碳氢键活化氧化反应(Chem. Eur. J. 2015, 21, 14937)、转移氢甲酰化反应(J. Org. Chem. 2016, 81, 2320.)、芳烃卤代反应(Chem. Eur. J. 2017, 23, 2690.)等。在这些研究中,反应机理可以总结为铑参与的碳氢键切断、碳铑键转化、催化剂再生等三个部分。蓝宇教授课题组的研究着眼于碳氢键切断方式、碳铑键转化过程中的化学选择性、催化循环中金属铑氧化态变化等科学问题,为铑催化碳氢键活化-官能团化反应机理提供了原理上的支持。
凭借以上系统性的研究工作,蓝宇教授课题组受美国化学会Accounts of Chemical Research杂志主编Burrows, Cynthia J.教授邀请撰写一篇题为《Mechanism of Rhodium-Catalyzed C−H Functionalization: Advances in Theoretical Investigation》的自我综述,以总结蓝宇教授课题组在铑催化碳氢键活化-官能团化反应机理研究的相关工作。最近,该论文发表在化学领域顶级期刊Accounts of Chemical Research杂志上。(Qi, X.; Li, Y.; Bai, R.; Lan, Y.* Acc. Chem. Res. 2017, 50, 2799-2808.(DOI:10.1021/acs.accounts.7b00400))该论文第一作者是重庆大学毕业的戚孝天博士;论文作者重庆大学博士研究生李英姿对该论文作出与第一作者同等贡献;论文作者还有重庆大学白若鹏博士;该论文通讯作者蓝宇教授,通讯地址为重庆大学。
Accounts of Chemical Research杂志在化学研究领域深具影响(2016年影响因子为20.27),意义在于了解现代化学的最新进展,被认为是化学化工领域顶级期刊之一,其发表的综述文章主要是描述作者自己的非常优秀的系统研究工作。
蓝宇教授课题组的研究工作是以密度泛函理论计算为主要研究方法,构建并使用基元反应路径形变-结合能模型研究均相催化反应机理相关问题,例如铑催化烃类官能团化反应、自由基参与成键反应等。在2014年以来的3年多里,蓝宇教授课题组以通讯作者身份在包括Accounts Chem. Res.(约稿1篇,IF = 20.27)、J. Am. Chem. Soc.(6篇,IF = 13.86)、Angew. Chem. Int. Ed.(9篇,IF = 11.99)、Nature Commun.(4篇,IF = 12.12)、Chem. Sci.(3篇,IF = 8.67)、ACS Catal.(5篇,IF = 10.61)、Sci. Adv.(1篇)、《中国科学:化学》(约稿3篇)等期刊发表科研论文共计80余篇(含已接收)。蓝宇教授通讯作者研究论文累计被他人正面引用达1000余次,全部论文累计被引用达2000余次。目前蓝宇教授课题组H因子为27(相关数据来源详见ResearcherID: A-8146-2016)。蓝宇教授自2014年起主持国家自然科学基金面上项目两项,青年项目1项。基于以上成绩蓝宇教授荣获“2016年中国化学会青年化学奖”。此外,蓝宇教授还荣获“2015年中国化学会-物理有机化学新人奖”。
论文概要:
Transition- l-catalyzed cross-coupling has emerged as an effective strategy for chemical synthesis. Within this area, direct C–H bond transformation is one of the most efficient and environmentally friendly processes for the construction of new C–C or C–heteroatom bonds. Over the past decades, rhodium-catalyzed C–H functionalization has attracted considerable attention because of the versatility and wide use of rhodium catalysts in chemistry. A series of C–X (X = C, N, or O) bond formation reactions could be realized from corresponding C–H bonds using rhodium catalysts. Various experimental studies on rhodium-catalyzed C–H functionalization reactions have been reported and in tandem, mechanistic and computational studies have also progressed significantly.
Since 2012, our group has performed theoretical studies to reveal the mechanism of rhodium-catalyzed C–H functionalization reactions. We have studied the changes in the oxidation state of rhodium and compared the Rh(I)/Rh(III) catalytic cycle to the Rh(III)/Rh(V) catalytic cycle using density functional theory calculation. The development of advanced computational methods and improvements in computing power make theoretical calculation a powerful tool for the mechanistic study of rhodium chemistry. Computational study is able to not only provide mechanistic insights, but also explain the origin of regioselectivity, enantioselectivity, and stereoselectivity in rhodium-catalyzed C–H functionalization reactions.
This account summarizes our computational work on rhodium-catalyzed C–H functionalization reactions. The mechanistic study under discussion is divided into three main parts: C–H bond cleavage step, transformation of the C–Rh bond, and regeneration of the active catalyst. In the C–H bond cleavage step, computational results of four possible mechanisms, including concerted lation deprotonation (CMD), oxidative addition (OA), Friedel-Crafts-type (SEAr), and σ-complex assisted thesis (σ-CAM) are discussed. Subsequent transformation of the C–Rh bond, for example, via insertion of CO, olefin, alkyne, carbene, or nitrene constructs new C–C or C–heteroatom bonds. For the regeneration of the active catalyst, reductive elimination of a high-valent rhodium complex and protonation of the C–Rh bond are emphasized as potential mechanism candidates. In addition to detailing the reaction pathway, the regioselectivity and diastereoselectivity of rhodium-catalyzed C–H functionalization reactions are also commented upon in this account. The origin of the selectivity is clarified through theoretical analysis. Furthermore, we summarize and compare the changes in the oxidation state of rhodium along the complete reaction pathway. The work described in this account demonstrates that rhodium catalysis might proceed via Rh(I)/Rh(III), Rh(II)/Rh(IV), Rh(III)/Rh(V), or non-redox-Rh(III) catalytic cycles.
